
AI2BMD
AI-powered ab initio biomolecular dynamics simulation
Stars: 155
AI2BMD is a program for efficiently simulating protein molecular dynamics with ab initio accuracy. The repository contains datasets, simulation programs, and public materials related to AI2BMD. It provides a Docker image for easy deployment and a standalone launcher program. Users can run simulations by downloading the launcher script and specifying simulation parameters. The repository also includes ready-to-use protein structures for testing. AI2BMD is designed for x86-64 GNU/Linux systems with recommended hardware specifications. The related research includes model architectures like ViSNet, Geoformer, and fine-grained force metrics for MLFF. Citation information and contact details for the AI2BMD Team are provided.
README:
- Overview
- Setup Guide
- Datasets
- System Requirements
- Related Research
- Citation
- License
- Disclaimer
- Contacts
AI2BMD is a program for efficiently simulating protein molecular dynamics with ab initio accuracy. This repository contains datasets, simulation programs, and public materials related to AI2BMD.
![]()
The source code of AI2BMD is hosted in this repository.
To streamline the user experience, we package the source code and runtime libraries into a Docker image, and provide a Python launcher program to simplify the deployment process.
To run the simulation program, you don't need to clone this repository. Simply download scripts/ai2bmd and launch it (Python >=3.7 is required).
wget 'https://raw.githubusercontent.com/microsoft/AI2BMD/main/scripts/ai2bmd'
chmod +x ai2bmd
# you may need to "sudo" the following line if the docker group is not configured for the user
./ai2bmd --prot-file path/to/target-protein.pdb --sim-steps nnn ...
# '-------- required argument ---------' '-- optional arguments --'
#
# Notable optional arguments:
#
# [Simulation directory mapping options]
# --base-dir path/to/base-dir A directory for running simulation (defaults to current directory)
# --log-dir path/to/log-dir A directory for saving results (defaults to base-dir/Logs-protein-name)
#
# [Simulation parameter options]
# --sim-steps nnn Simulation steps
# --temp-k nnn Simulation temperature in Kelvin
# --timestep nnn TimeStep (fs) for simulation
# --preeq-steps nnn Pre-equilibration simulation steps for each constraint
# --max-cyc nnn Maximum energy minimization cycles in preprocessing
#
# [Performance tweaks]
# --device-strategy [strategy] The compute device allocation strategy
# small-molecule Bonded/non-bonded/solvent computation share all GPUs, enable GPU oversubscription
# large-molecule No multiple models on the same GPU
# --chunk-size nnn When there's more than device_chunk elements (e.g. dipeptides) in a batch, split them into chunks
# and feed them into GPUs sequentially. Reduces memory consumption
#
# [Additional launcher options]
# --software-update When specified, updates the program in the Docker image before running
# --download-training-data When specified, downloads the AI2BMD training data, and unpacks it in the working directory.
# Ignores all other options.
# --gpus Specifies the GPU devices to passthrough to the program. Can be one of the following:
# all: Passthrough all available GPUs to the program.
# none: Disables GPU passthrough.
# i[,j,k...] Passthrough some GPUs. Example: --gpus 0,1The code repository contains several sample protein structures in the testcases directory. Here we use the Chignolin structure as an example:
# skip the following two lines if you've already set up the launcher
wget 'https://raw.githubusercontent.com/microsoft/AI2BMD/main/scripts/ai2bmd'
chmod +x ai2bmd
# download the Chignolin protein structure data file
wget 'https://raw.githubusercontent.com/microsoft/AI2BMD/main/testcases/chig.pdb'
# launch the program, with all simulation parameters set to default values
# you may need to "sudo" the following line if the docker group is not configured for the user
./ai2bmd --prot-file chig.pdbThe results will be placed in a new directory Logs-chig.
The directory contains the simulation trajectory file:
- chig-traj.traj: The full trajectory file in ASE binary format.
The protein unit dataset covers a wide range of conformations for dipeptides. It can be downloaded with the following commands:
# skip the following two lines if you've already set up the launcher
wget 'https://raw.githubusercontent.com/microsoft/AI2BMD/main/scripts/ai2bmd'
chmod +x ai2bmd
# you may need to "sudo" the following line if the docker group is not configured for the user
./ai2bmd --download-training-dataWhen it finishes, the current working directory will be populated by the numpy data files (*.npz).
The whole comformation MD dataset for proteins calculated at Density Functional Theory (DFT) level. AIMD-Chig consists of 2M conformations of the 166-atom Chignolin and the corresponding potential energy and atomic forces calculated at M06-2X/6-31g* level.
-
Read the article AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics.
-
Find the story The first whole conformational molecular dynamics dataset for proteins at ab initio accuracy and the novel computational technologies behind it.
-
Get the dataset AIMD-Chig.
The AI2BMD program runs on x86-64 GNU/Linux systems. We recommend a machine with the following specs:
- CPU: 8+ cores
- Memory: 32+ GB
- GPU: CUDA-enabled GPU with 8+ GB memory
The program has been tested on the following GPUs:
- A100
- V100
- RTX A6000
- Titan RTX
The program has been tested on the following systems:
- OS: Ubuntu 20.04, Docker: 27.1
- OS: ArchLinux, Docker: 26.1
ViSNet (Vector-Scalar interactive graph neural Network) is an equivariant geometry-enhanced graph neural for molecules that significantly alleviates the dilemma between computational costs and the sufficient utilization of geometric information.
-
ViSNet is published on Nature Communications Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing.
-
ViSNet is selected as "Editors' Highlights" for both "AI and machine learning" and "Biotechnology and methods" fields of Nature Communications.
-
ViSNet has won the Championship in The First Global AI Drug Development Competition and one of the winners in OGB-LSC @ NeurIPS 2022 PCQM4Mv2 Track!
-
Please check out the branch ViSNet for the source code, instructions on model training, and more techniqucal details.
Geoformer (Geometric Transformer) is a novel geometric Transformer to effectively model molecular structures for various molecular property predictions. Geoformer introduces a novel positional encoding method, Interatomic Positional Encoding (IPE), to parameterize atomic environments in Transformer. By incorporating IPE, Geoformer captures valuable geometric information beyond pairwise distances within a Transformer-based architecture. Geoformer can be regarded as a Transformer variant of ViSNet.
- Geoformer was published on NeurIPS 2023.
- Read the paper of Geoformer Geometric Transformer with Interatomic Positional Encoding.
- Please check out the branch Geoformer for the source code, instructions on model training, and more techniqucal details.
Machine learning force fields (MLFFs) have gained popularity in recent years as a cost-effective alternative to ab initio molecular dynamics (MD) simulations. Despite their small errors on test sets, MLFFs inherently suffer from generalization and robustness issues during MD simulations.
To alleviate these issues, we propose the use of global force metrics and fine-grained metrics from elemental and conformational aspects to systematically measure MLFFs for every atom and conformation of molecules. Furthermore, the performance of MLFFs and the stability of MD simulations can be enhanced by employing the proposed force metrics during model training. This includes training MLFF models using these force metrics as loss functions, fine-tuning by reweighting samples in the original dataset, and continued training by incorporating additional unexplored data.
- Read the Cover Story article Improving machine learning force fields for molecular dynamics simulations with fine-grained force metrics .
Markov state models (MSMs) play a key role in studying protein conformational dynamics. A sliding count window with a fixed lag time is commonly used to sample sub-trajectories for transition counting and MSM construction. However, sub-trajectories sampled with a fixed lag time may not perform well under different selections of lag time, requiring strong prior experience and resulting in less robust estimations.
To alleviate this, we propose a novel stochastic method based on a Poisson process to generate perturbative lag times for sub-trajectory sampling and use it to construct a Markov chain. Comprehensive evaluations on the double-well system, WW domain, BPTI, and RBD–ACE2 complex of SARS-CoV-2 reveal that our algorithm significantly increases the robustness and accuracy of the constructed MSM without disrupting its Markovian properties. Furthermore, the advantages of our algorithm are especially pronounced for slow dynamic modes in complex biological processes.
-
Read the Cover Story article Stochastic Lag Time Parameterization for Markov State Models of Protein Dynamics.
-
Find an application case in studying the Spike-ACE2 complex structure for the highly infectious mechanism of Omicron: Structural insights into the SARS-CoV-2 Omicron RBD-ACE2 interaction.
(#: co-first author; *: corresponding author)
Yusong Wang#, Tong Wang#*, Shaoning Li#, Xinheng He, Mingyu Li, Zun Wang, Nanning Zheng, Bin Shao*, Tie-Yan Liu, Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing, Nature Communications, 15.1 (2024): 313.
Yusong Wang#, Shaoning Li#, Tong Wang*, Bin Shao, Nanning Zheng, Tie-Yan Liu. Geometric Transformer with Interatomic Positional Encoding. NeurIPS 2023.
Zun Wang#, Hongfei Wu#, Lixin Sun, Xinheng He, Zhirong Liu, Bin Shao, Tong Wang*, Tie-Yan Liu. Improving machine learning force fields for molecular dynamics simulations with fine-grained force metrics, The Journal of Chemical Physics, Volume 159, Issue 3, Cover Story.
Tong Wang#*, Xinheng He#, Mingyu Li#, Bin Shao*, Tie-Yan Liu. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics, Scientific Data 10, 549 (2023).
Shiqi Gong#, Xinheng He#, Qi Meng, Zhiming Ma, Bin Shao*, Tong Wang*, Tie-Yan Liu. Stochastic Lag Time Parameterization for Markov State Models of Protein Dynamics, The Journal of Physical Chemistry B 2022 126 (46), Cover Story, 2022.
Copyright (c) Microsoft Corporation. All rights reserved.
Licensed under the MIT license.
AI2BMD is a research project. It is not an officially supported Microsoft product.
Please contact AI2BMD Team for any questions or suggestions. The main team members include:
- Tong Wang (Primary lead/contact)
- Yatao Li
- Ran Bi
- Bin Shao
- Tie-Yan Liu
For Tasks:
Click tags to check more tools for each tasksFor Jobs:
Alternative AI tools for AI2BMD
Similar Open Source Tools
AI2BMD
AI2BMD is a program for efficiently simulating protein molecular dynamics with ab initio accuracy. The repository contains datasets, simulation programs, and public materials related to AI2BMD. It provides a Docker image for easy deployment and a standalone launcher program. Users can run simulations by downloading the launcher script and specifying simulation parameters. The repository also includes ready-to-use protein structures for testing. AI2BMD is designed for x86-64 GNU/Linux systems with recommended hardware specifications. The related research includes model architectures like ViSNet, Geoformer, and fine-grained force metrics for MLFF. Citation information and contact details for the AI2BMD Team are provided.
BitMat
BitMat is a Python package designed to optimize matrix multiplication operations by utilizing custom kernels written in Triton. It leverages the principles outlined in the "1bit-LLM Era" paper, specifically utilizing packed int8 data to enhance computational efficiency and performance in deep learning and numerical computing tasks.
RAGEN
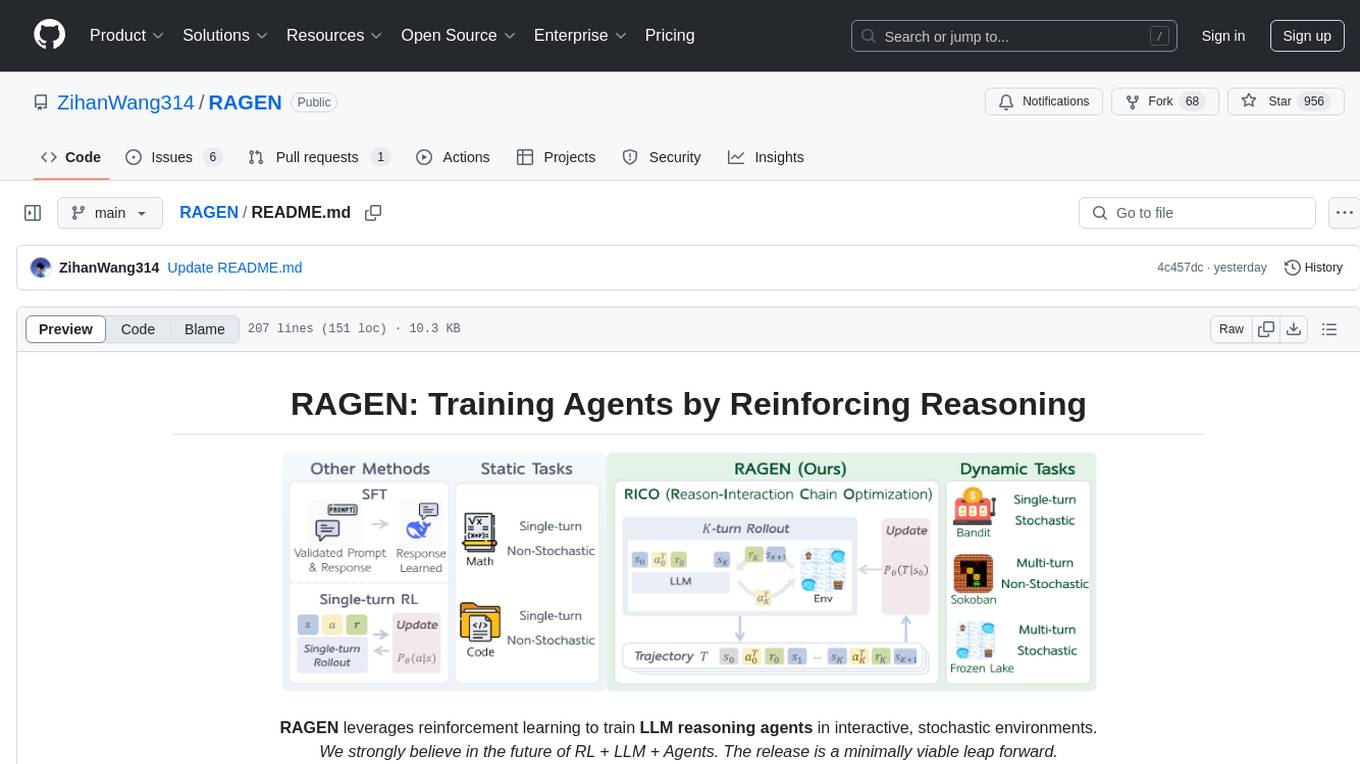
RAGEN is a reinforcement learning framework designed to train reasoning-capable large language model (LLM) agents in interactive, stochastic environments. It addresses challenges such as multi-turn interactions and stochastic environments through a Markov Decision Process (MDP) formulation, Reason-Interaction Chain Optimization (RICO) algorithm, and progressive reward normalization strategies. The framework enables LLMs to reason and interact with the environment, optimizing entire trajectories for long-horizon reasoning while maintaining computational efficiency.
RAGEN
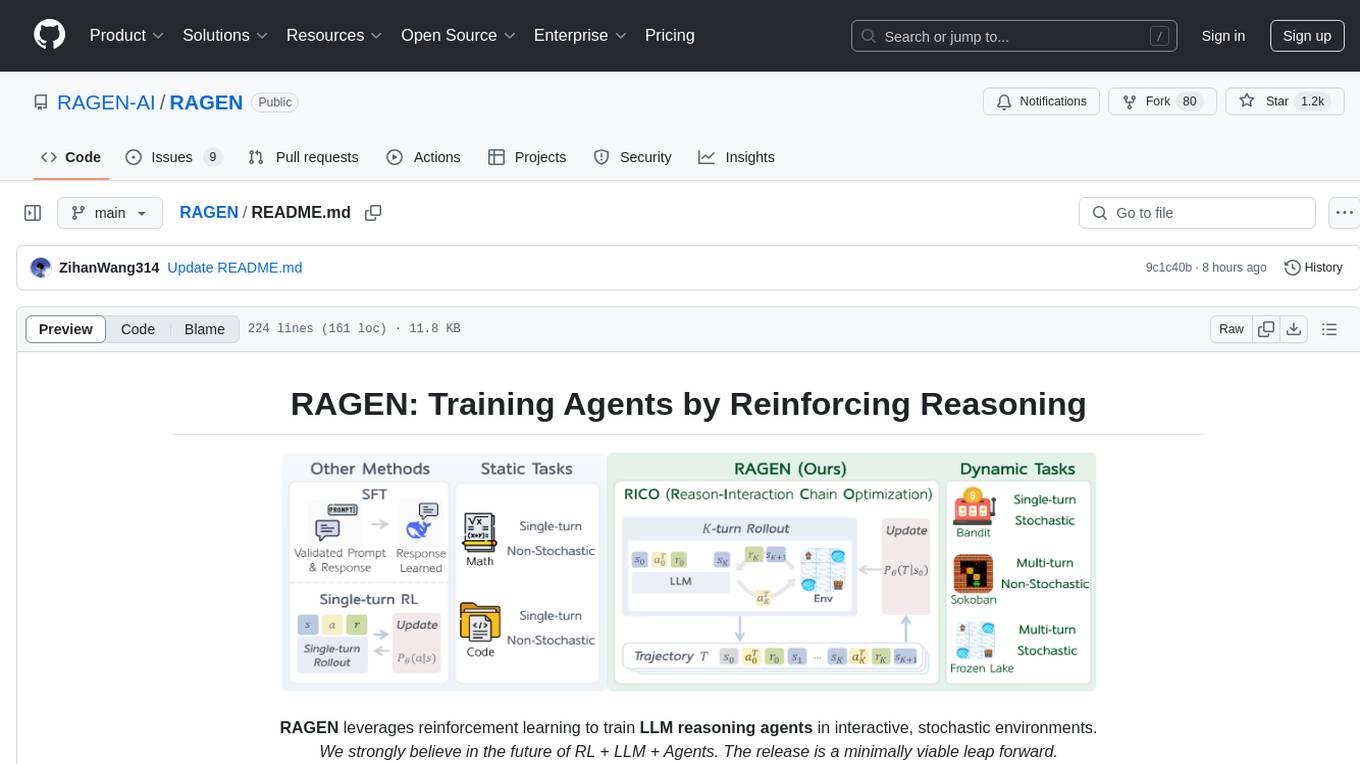
RAGEN is a reinforcement learning framework designed to train reasoning-capable large language model (LLM) agents in interactive, stochastic environments. It addresses challenges such as multi-turn interactions and stochastic environments through a Markov Decision Process (MDP) formulation, Reason-Interaction Chain Optimization (RICO) algorithm, and progressive reward normalization strategies. The framework consists of MDP formulation, RICO algorithm with rollout and update stages, and reward normalization strategies to stabilize training. RAGEN aims to optimize reasoning and action strategies for LLM agents operating in complex environments.
aligner
Aligner is a model-agnostic alignment tool that learns correctional residuals between preferred and dispreferred answers using a small model. It can be directly applied to various open-source and API-based models with only one-off training, suitable for rapid iteration and improving model performance. Aligner has shown significant improvements in helpfulness, harmlessness, and honesty dimensions across different large language models.

ReaLHF
ReaLHF is a distributed system designed for efficient RLHF training with Large Language Models (LLMs). It introduces a novel approach called parameter reallocation to dynamically redistribute LLM parameters across the cluster, optimizing allocations and parallelism for each computation workload. ReaL minimizes redundant communication while maximizing GPU utilization, achieving significantly higher Proximal Policy Optimization (PPO) training throughput compared to other systems. It supports large-scale training with various parallelism strategies and enables memory-efficient training with parameter and optimizer offloading. The system seamlessly integrates with HuggingFace checkpoints and inference frameworks, allowing for easy launching of local or distributed experiments. ReaLHF offers flexibility through versatile configuration customization and supports various RLHF algorithms, including DPO, PPO, RAFT, and more, while allowing the addition of custom algorithms for high efficiency.
llm-analysis
llm-analysis is a tool designed for Latency and Memory Analysis of Transformer Models for Training and Inference. It automates the calculation of training or inference latency and memory usage for Large Language Models (LLMs) or Transformers based on specified model, GPU, data type, and parallelism configurations. The tool helps users to experiment with different setups theoretically, understand system performance, and optimize training/inference scenarios. It supports various parallelism schemes, communication methods, activation recomputation options, data types, and fine-tuning strategies. Users can integrate llm-analysis in their code using the `LLMAnalysis` class or use the provided entry point functions for command line interface. The tool provides lower-bound estimations of memory usage and latency, and aims to assist in achieving feasible and optimal setups for training or inference.
HuixiangDou2
HuixiangDou2 is a robustly optimized GraphRAG approach that integrates multiple open-source projects to improve performance in graph-based augmented generation. It conducts comparative experiments and achieves a significant score increase, leading to a GraphRAG implementation with recognized performance. The repository provides code improvements, dense retrieval for querying entities and relationships, real domain knowledge testing, and impact analysis on accuracy.
nixtla
Nixtla is a production-ready generative pretrained transformer for time series forecasting and anomaly detection. It can accurately predict various domains such as retail, electricity, finance, and IoT with just a few lines of code. TimeGPT introduces a paradigm shift with its standout performance, efficiency, and simplicity, making it accessible even to users with minimal coding experience. The model is based on self-attention and is independently trained on a vast time series dataset to minimize forecasting error. It offers features like zero-shot inference, fine-tuning, API access, adding exogenous variables, multiple series forecasting, custom loss function, cross-validation, prediction intervals, and handling irregular timestamps.
pytorch-forecasting
PyTorch Forecasting is a PyTorch-based package for time series forecasting with state-of-the-art network architectures. It offers a high-level API for training networks on pandas data frames and utilizes PyTorch Lightning for scalable training on GPUs and CPUs. The package aims to simplify time series forecasting with neural networks by providing a flexible API for professionals and default settings for beginners. It includes a timeseries dataset class, base model class, multiple neural network architectures, multi-horizon timeseries metrics, and hyperparameter tuning with optuna. PyTorch Forecasting is built on pytorch-lightning for easy training on various hardware configurations.
Controllable-RAG-Agent
This repository contains a sophisticated deterministic graph-based solution for answering complex questions using a controllable autonomous agent. The solution is designed to ensure that answers are solely based on the provided data, avoiding hallucinations. It involves various steps such as PDF loading, text preprocessing, summarization, database creation, encoding, and utilizing large language models. The algorithm follows a detailed workflow involving planning, retrieval, answering, replanning, content distillation, and performance evaluation. Heuristics and techniques implemented focus on content encoding, anonymizing questions, task breakdown, content distillation, chain of thought answering, verification, and model performance evaluation.
Nanoflow
NanoFlow is a throughput-oriented high-performance serving framework for Large Language Models (LLMs) that consistently delivers superior throughput compared to other frameworks by utilizing key techniques such as intra-device parallelism, asynchronous CPU scheduling, and SSD offloading. The framework proposes nano-batching to schedule compute-, memory-, and network-bound operations for simultaneous execution, leading to increased resource utilization. NanoFlow also adopts an asynchronous control flow to optimize CPU overhead and eagerly offloads KV-Cache to SSDs for multi-round conversations. The open-source codebase integrates state-of-the-art kernel libraries and provides necessary scripts for environment setup and experiment reproduction.
dash-infer
DashInfer is a C++ runtime tool designed to deliver production-level implementations highly optimized for various hardware architectures, including x86 and ARMv9. It supports Continuous Batching and NUMA-Aware capabilities for CPU, and can fully utilize modern server-grade CPUs to host large language models (LLMs) up to 14B in size. With lightweight architecture, high precision, support for mainstream open-source LLMs, post-training quantization, optimized computation kernels, NUMA-aware design, and multi-language API interfaces, DashInfer provides a versatile solution for efficient inference tasks. It supports x86 CPUs with AVX2 instruction set and ARMv9 CPUs with SVE instruction set, along with various data types like FP32, BF16, and InstantQuant. DashInfer also offers single-NUMA and multi-NUMA architectures for model inference, with detailed performance tests and inference accuracy evaluations available. The tool is supported on mainstream Linux server operating systems and provides documentation and examples for easy integration and usage.
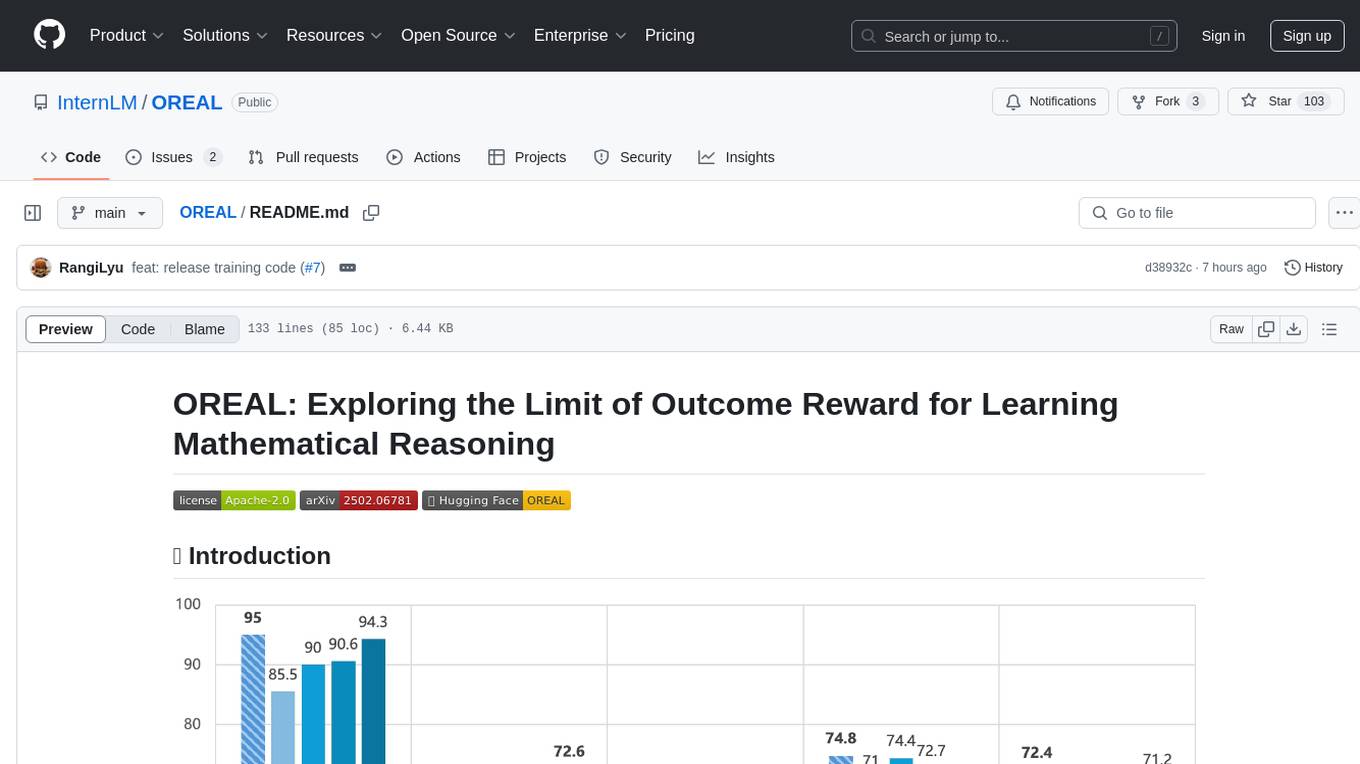
OREAL
OREAL is a reinforcement learning framework designed for mathematical reasoning tasks, aiming to achieve optimal performance through outcome reward-based learning. The framework utilizes behavior cloning, reshaping rewards, and token-level reward models to address challenges in sparse rewards and partial correctness. OREAL has achieved significant results, with a 7B model reaching 94.0 pass@1 accuracy on MATH-500 and surpassing previous 32B models. The tool provides training tutorials and Hugging Face model repositories for easy access and implementation.
AlphaFold3
AlphaFold3 is an implementation of the Alpha Fold 3 model in PyTorch for accurate structure prediction of biomolecular interactions. It includes modules for genetic diffusion and full model examples for forward pass computations. The tool allows users to generate random pair and single representations, operate on atomic coordinates, and perform structure predictions based on input tensors. The implementation also provides functionalities for training and evaluating the model.
GOLEM
GOLEM is an open-source AI framework focused on optimization and learning of structured graph-based models using meta-heuristic methods. It emphasizes the potential of meta-heuristics in complex problem spaces where gradient-based methods are not suitable, and the importance of structured models in various problem domains. The framework offers features like structured model optimization, metaheuristic methods, multi-objective optimization, constrained optimization, extensibility, interpretability, and reproducibility. It can be applied to optimization problems represented as directed graphs with defined fitness functions. GOLEM has applications in areas like AutoML, Bayesian network structure search, differential equation discovery, geometric design, and neural architecture search. The project structure includes packages for core functionalities, adapters, graph representation, optimizers, genetic algorithms, utilities, serialization, visualization, examples, and testing. Contributions are welcome, and the project is supported by ITMO University's Research Center Strong Artificial Intelligence in Industry.
For similar tasks
AI2BMD
AI2BMD is a program for efficiently simulating protein molecular dynamics with ab initio accuracy. The repository contains datasets, simulation programs, and public materials related to AI2BMD. It provides a Docker image for easy deployment and a standalone launcher program. Users can run simulations by downloading the launcher script and specifying simulation parameters. The repository also includes ready-to-use protein structures for testing. AI2BMD is designed for x86-64 GNU/Linux systems with recommended hardware specifications. The related research includes model architectures like ViSNet, Geoformer, and fine-grained force metrics for MLFF. Citation information and contact details for the AI2BMD Team are provided.
AI-Drug-Discovery-Design
AI-Drug-Discovery-Design is a repository focused on Artificial Intelligence-assisted Drug Discovery and Design. It explores the use of AI technology to accelerate and optimize the drug development process. The advantages of AI in drug design include speeding up research cycles, improving accuracy through data-driven models, reducing costs by minimizing experimental redundancies, and enabling personalized drug design for specific patients or disease characteristics.
AIRS
AIRS is a collection of open-source software tools, datasets, and benchmarks focused on Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems. The goal is to develop and maintain an integrated, open, reproducible, and sustainable set of resources to advance the field of AI for Science. The current resources include tools for Quantum Mechanics, Density Functional Theory, Small Molecules, Protein Science, Materials Science, Molecular Interactions, and Partial Differential Equations.
Magick
Magick is a groundbreaking visual AIDE (Artificial Intelligence Development Environment) for no-code data pipelines and multimodal agents. Magick can connect to other services and comes with nodes and templates well-suited for intelligent agents, chatbots, complex reasoning systems and realistic characters.

metaflow
Metaflow is a user-friendly library designed to assist scientists and engineers in developing and managing real-world data science projects. Initially created at Netflix, Metaflow aimed to enhance the productivity of data scientists working on diverse projects ranging from traditional statistics to cutting-edge deep learning. For further information, refer to Metaflow's website and documentation.
SciMLBenchmarks.jl
SciMLBenchmarks.jl holds webpages, pdfs, and notebooks showing the benchmarks for the SciML Scientific Machine Learning Software ecosystem, including: * Benchmarks of equation solver implementations * Speed and robustness comparisons of methods for parameter estimation / inverse problems * Training universal differential equations (and subsets like neural ODEs) * Training of physics-informed neural networks (PINNs) * Surrogate comparisons, including radial basis functions, neural operators (DeepONets, Fourier Neural Operators), and more The SciML Bench suite is made to be a comprehensive open source benchmark from the ground up, covering the methods of computational science and scientific computing all the way to AI for science.

katib
Katib is a Kubernetes-native project for automated machine learning (AutoML). Katib supports Hyperparameter Tuning, Early Stopping and Neural Architecture Search. Katib is the project which is agnostic to machine learning (ML) frameworks. It can tune hyperparameters of applications written in any language of the users’ choice and natively supports many ML frameworks, such as TensorFlow, Apache MXNet, PyTorch, XGBoost, and others. Katib can perform training jobs using any Kubernetes Custom Resources with out of the box support for Kubeflow Training Operator, Argo Workflows, Tekton Pipelines and many more.
X-AnyLabeling
X-AnyLabeling is a robust annotation tool that seamlessly incorporates an AI inference engine alongside an array of sophisticated features. Tailored for practical applications, it is committed to delivering comprehensive, industrial-grade solutions for image data engineers. This tool excels in swiftly and automatically executing annotations across diverse and intricate tasks.
For similar jobs
Generative-AI-Drug-Discovery
Generative-AI-Drug-Discovery is a public repository on GitHub focused on using tensor network machine learning approaches to accelerate GenAI for drug discovery. The repository aims to implement effective architectures and methodologies into Large Language Models (LLMs) to enhance Drug Discovery Generative AI performance.
AI2BMD
AI2BMD is a program for efficiently simulating protein molecular dynamics with ab initio accuracy. The repository contains datasets, simulation programs, and public materials related to AI2BMD. It provides a Docker image for easy deployment and a standalone launcher program. Users can run simulations by downloading the launcher script and specifying simulation parameters. The repository also includes ready-to-use protein structures for testing. AI2BMD is designed for x86-64 GNU/Linux systems with recommended hardware specifications. The related research includes model architectures like ViSNet, Geoformer, and fine-grained force metrics for MLFF. Citation information and contact details for the AI2BMD Team are provided.
mercure
mercure DICOM Orchestrator is a flexible solution for routing and processing DICOM files. It offers a user-friendly web interface and extensive monitoring functions. Custom processing modules can be implemented as Docker containers. Written in Python, it uses the DCMTK toolkit for DICOM communication. It can be deployed as a single-server installation using Docker Compose or as a scalable cluster installation using Nomad. mercure consists of service modules for receiving, routing, processing, dispatching, cleaning, web interface, and central monitoring.
ProLLM
ProLLM is a framework that leverages Large Language Models to interpret and analyze protein sequences and interactions through natural language processing. It introduces the Protein Chain of Thought (ProCoT) method to transform complex protein interaction data into intuitive prompts, enhancing predictive accuracy by incorporating protein-specific embeddings and fine-tuning on domain-specific datasets.
grand-challenge.org
Grand Challenge is a platform that provides access to large amounts of annotated training data, objective comparisons of state-of-the-art machine learning solutions, and clinical validation using real-world data. It assists researchers, data scientists, and clinicians in collaborating to develop robust machine learning solutions to problems in biomedical imaging.
Detection-and-Classification-of-Alzheimers-Disease
This tool is designed to detect and classify Alzheimer's Disease using Deep Learning and Machine Learning algorithms on an early basis, which is further optimized using the Crow Search Algorithm (CSA). Alzheimer's is a fatal disease, and early detection is crucial for patients to predetermine their condition and prevent its progression. By analyzing MRI scanned images using Artificial Intelligence technology, this tool can classify patients who may or may not develop AD in the future. The CSA algorithm, combined with ML algorithms, has proven to be the most effective approach for this purpose.
OpenCRISPR
OpenCRISPR is a set of free and open gene editing systems designed by Profluent Bio. The OpenCRISPR-1 protein maintains the prototypical architecture of a Type II Cas9 nuclease but is hundreds of mutations away from SpCas9 or any other known natural CRISPR-associated protein. You can view OpenCRISPR-1 as a drop-in replacement for many protocols that need a cas9-like protein with an NGG PAM and you can even use it with canonical SpCas9 gRNAs. OpenCRISPR-1 can be fused in a deactivated or nickase format for next generation gene editing techniques like base, prime, or epigenome editing.
AlphaFold3
AlphaFold3 is an implementation of the Alpha Fold 3 model in PyTorch for accurate structure prediction of biomolecular interactions. It includes modules for genetic diffusion and full model examples for forward pass computations. The tool allows users to generate random pair and single representations, operate on atomic coordinates, and perform structure predictions based on input tensors. The implementation also provides functionalities for training and evaluating the model.